Institut für Zellbiologie
Das zentrale Anliegen der Zellbiologie ist die Erforschung der Lebensvorgänge auf zellulärer Ebene. Der Fokus hat sich hierbei immer mehr von einer rein morphologischen Beobachtung auf die Analyse molekularer Regulationsmechanismen verlagert. Unsere Forschung liefert wichtige Grundlagen für das Verstehen von Krankheitsmechanismen und ist das Fundament für die Entwicklung möglicher Therapieansätze.
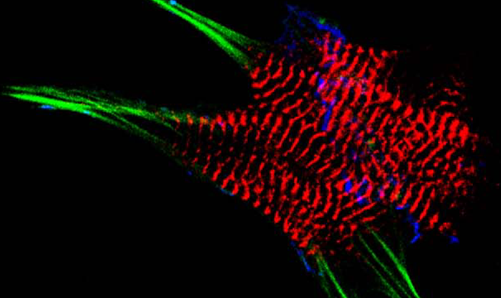
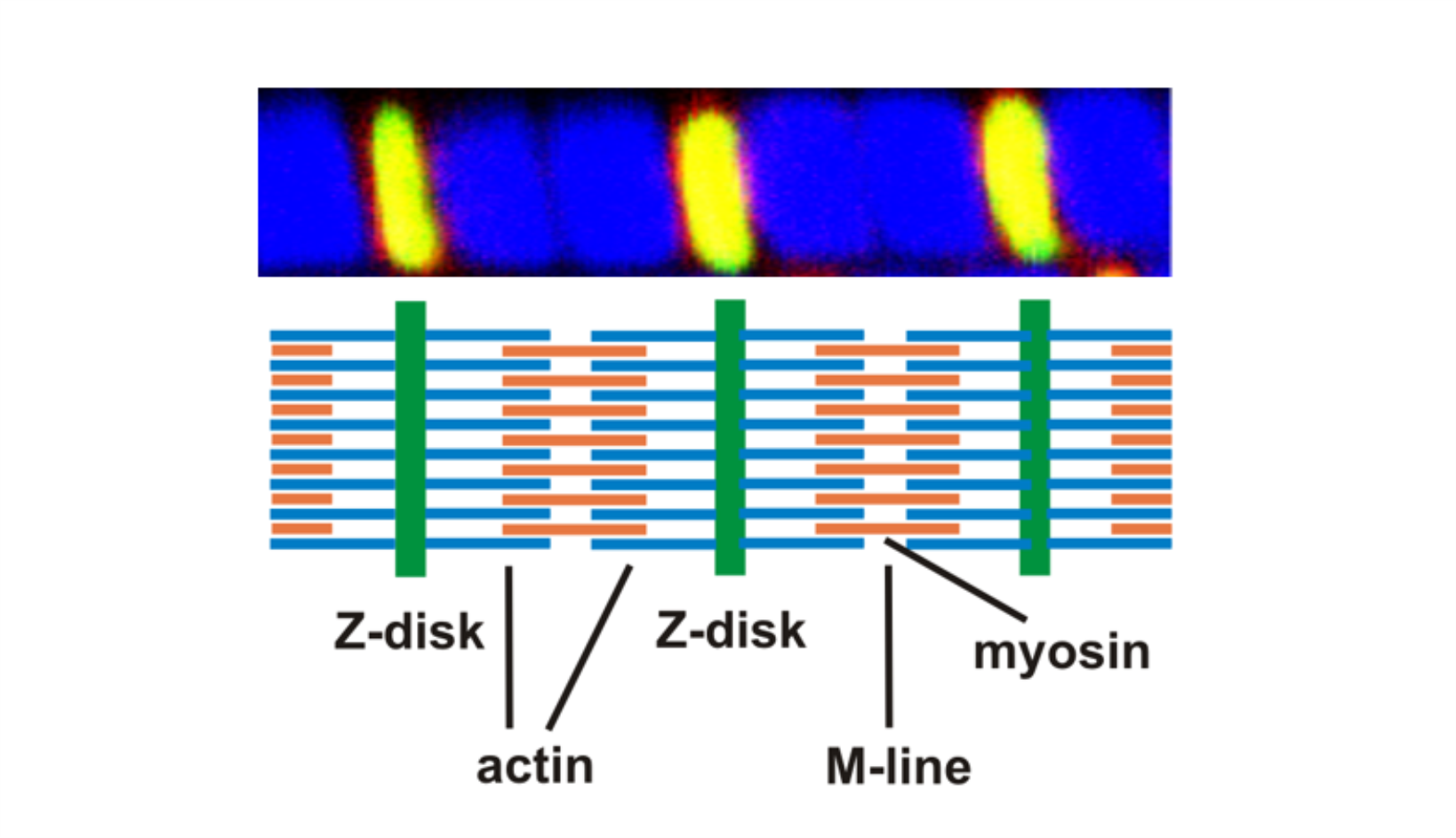
AG Fürst: Molekulare Zellbiologie des Muskels
Welche Protein-Protein Wechselwirkungen sind die molekulare Basis für den Aufbau komplexer biologischer Strukturen? Welche Mechanismen sorgen für den Erhalt dieser Strukturen unter Stress? Welche Fehlsteuerungen sind an pathophysiologischen Prozessen beteiligt?

AG Haas: Molekulare Zellbiologie der Infektion
Was geschieht in einer Immunfresszelle, nachdem Sie ein Partikel aufgenommen hat und wie programmieren pathogene Bakterien diesen Prozess um?
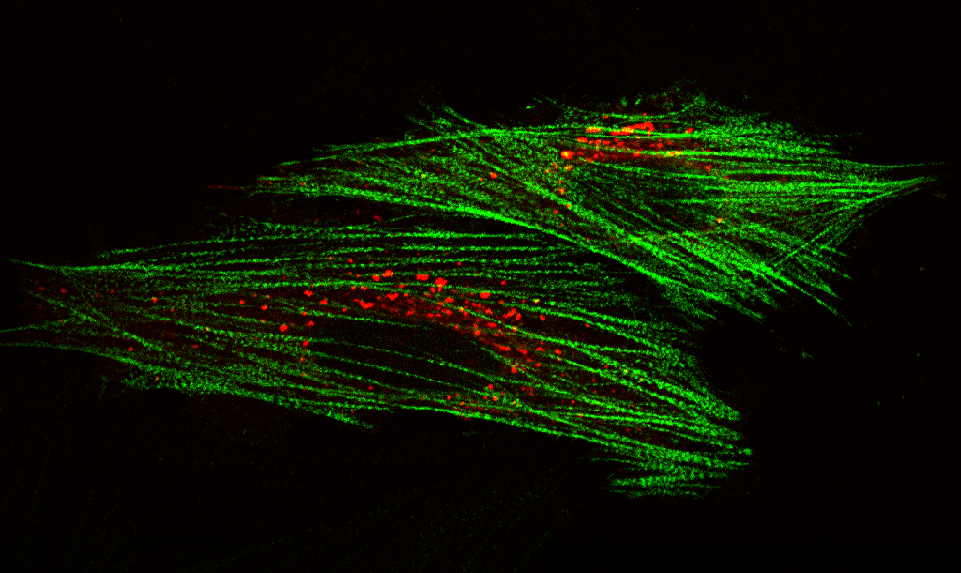
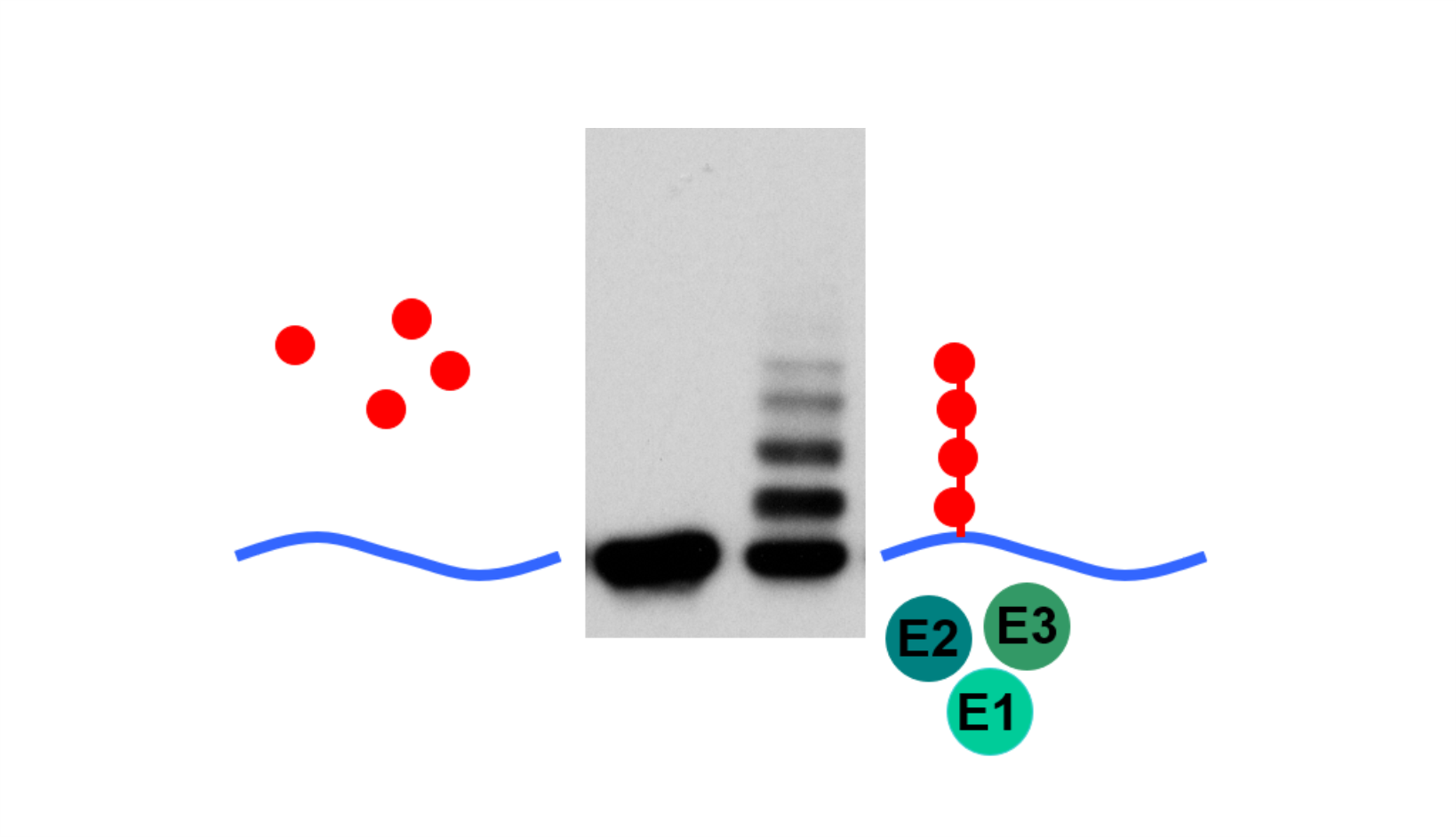
AG Höhfeld: Molekulare Zellbiologie der Proteom-Erhaltung
Wie wird die Proteinausstattung unserer Zellen und Gewebe aufrechterhalten und wie beeinflussen Erkrankungen und das Altern die Proteom-Erhaltung?
Mögliche Themen für Abschlussarbeiten oder Forschungspraktika finden Sie auf den Seiten der beiden Arbeitsgruppen.
Lehre Bachelor Biologie
(inkl. Lehramt)
Wir sind maßgeblich an der Lehre im Studiengang Biologie beteiligt.
Lehre für Master-Studierende
(MCB)
Wir vermitteln eine moderne Zellbiologie in verschiedenen Master-Studiengängen der Universität Bonn.

Ansprechpartner
& allgemeine Organisation
Wir freuen uns über Ihre Kontaktaufnahme.
Kontakt
Geschäftszimmer: Sabine Scharfenberg
Leiter: Prof. Dr. Dieter Fürst
Adresse
1. Etage, Raum 1.012
Ulrich-Haberland-Str. 61a
53121 Bonn